Bioinformatika - úvod
Jedným z charakteristických znakov moderného genomického výskumu je generovanie obrovského množstva surových sekvenčných údajov. S rastúcim objemom genomických údajov sú na zvládnutie veľkého množsta dát potrebné sofistikované výpočtové metodológie. Úplne prvou výzvou v ére genomiky je teda uchovávať a spracovávať ohromujúci objem informácií prostredníctvom vytvárania a používania počítačových databáz.
Hlavným cieľom bioinformatiky je prepojenie všetkých známych genetických, molekulárnych či biologických informácií, ktoré by mali slúžiť k poskytnutiu nových poznatkov, ktoré si môžeme všimnúť len vtedy, ak dokážeme vidieť celkový obraz. Bioinformatika nám umožňuje zhromaždiť údaje z mnohých experimentov na jednom mieste.
Aplikácia výpočtových techník na analýzu informácií spojených s biomolekulami sa rozvinula ako disciplína molekulárnej biológie a zahŕňa širokú škálu tematických oblastí od štrukturálnej biológie, genomiky až po štúdie génovej expresie.
Vo všeobecnosti má bioinformatika tieto základné ciele:
• Tvorba a správa databáz biologických dát rôzneho charakteru (DNA, RNA alebo proteínové sekvencie, proteínové štruktúry, génové expresné profily, atď.)
• Tvorba algoritmov a matematických modelov určených na analýzu biologických dát
• Aplikácia bioinformatických nástrojov za účelom interpretácie týchto dát vo svetle biologických súvislostí
Bioinformatika - história
Pre súčasných študentov a výskumníkov sa môže moderná bioinformatika javiť ako relatívne nedávna veda, ktorá prichádza na záchranu analýzy údajov NGS. Samotné začiatky bioinformatiky však nastali pred viac ako 50 rokmi, keď boli stolné počítače ešte len hypotézou a DNA sa ešte nedala sekvenovať.
1950-1970: Začiatkom 50. rokov minulého storočia sa o deoxyribonukleovej kyseline (DNA) veľa nevedelo. Jeho postavenie ako nosnej molekuly genetickej informácie bolo v tom čase stále kontroverzné. Predpokladalo sa, že nositeľom genetickej informácie sú proteíny. Koncom 50. rokov 20. storočia bola publikovaná prvá sekvencia proteínu – inzulínu.
1965: Margaret Dayhoffová vyvinula prvú databázu proteínových sekvencií, ktorá sa nazývala Atlas proteínovej sekvencie a štruktúry. To bol hlavný krok k pochopeniu vzťahu medzi štruktúrou a funkciou proteínu.
1970: Walter M. Fitch, zadefinoval pojem ortológie. Ortológy sú zadefinované ako gény, ktoré sa vyvinuli zo spoločného rodového génu speciáciou, ktoré si zvyčajne zachovali podobnú funkciu u rôznych druhov . Na druhej strane, paralógy sú gény súvisiace duplikáciou v rámci genómu a často získavajú novú funkciu. Napríklad bolop zistené, že ľudský hemoglobín vykazuje vyššiu konzerváciu so šimpanzím ( Pan troglodytes ) hemoglobínom ako s myším (Mus musculus) hemoglobínom. Ortologické proteíny sa teda vyvinuli prostredníctvom divergencie od spoločného predka.

Sekvenčná odlišnosť medzi ortologickými proteínmi z modelových organizmov koreluje s ich evolučnou históriou, ako to dokazujú i fosílne záznamy. ( A ) Priemerná vzdialenosť stromu hemoglobínovej podjednotky beta-1 (HBB-1) od človeka ( Homo sapiens ), šimpanza ( Pan troglodytes ), potkana ( Rattus norvegicus ), kurčaťa ( Gallus gallus ) a zebričky ( Danio rerio ). ( B ) Pohľad na porovnanie prvých 14 aminokyselinových zvyškov HBB-1 v porovnaní s ( A) (zvyšky zvýraznené modrou farbou sú identické so sekvenciou ľudského HBB-1). ( C ) Časová os najskorších fosílií nájdených pre rôzne vodné a suchozemské živočíchy (ZDROJ: Brief Bioinform, Volume 20, Issue 6, November 2019, Pages 1981–1996, https://doi.org/10.1093/bib/bby063).
1971:PDB Proteínová databanka s obsahom 7 proteinových štruktúr, RCSB (Research Collaboratory for Structural Bioinformatics)1981: Smithov-Watermanov algoritmus na zarovnanie sekvencií, užitočný pri identifikácii oblastí podobnosti, ktoré môžu naznačovať funkčné, štrukturálne alebo evolučné vzťahy medzi dvoma sekvenciami.
1982: GenBank, databáza nukleotidových sekvencií, vytvorená Národným inštitútom zdravia (NIH) ako spôsob ukladania a zdieľania genetických informácií.
1988: Vývoj softvérových nástrojov FASTA a BLAST slúžiacich na vyhľadávanie podobností, ktoré identifikujú homológne sekvencie DNA a proteínov na základe nadmernej podobnosti sekvencií. Poskytujú možnosti na porovnávanie sekvencií DNA a proteínov s existujúcimi databázami DNA a proteínov.
1990: Bol spustený projekt ľudského genómu. Cieľom tohto ambiciózneho projektu bolo sekvenovať celý ľudský genóm a bol dokončený v roku 2003.
1996: Bola vytvorená prvá proteomická databáza SWISS-PROT. Táto databáza obsahuje informácie o proteínových sekvenciách, funkciách a štruktúrach.
2001: Bol zverejnený prvý návrh ľudského genómu. To bol veľký prelom v našom chápaní biológie človeka a otvorilo to nové cesty pre výskum a vývoj liekov.
2002: Databáza proteínových sekvencií UniProt
2012: Bol objavený systém CRISPR-Cas9. Táto revolučná technológia umožňuje vedcom upravovať genómy s bezprecedentnou presnosťou.
2023: Integrácia umelej inteligencie (AI) a strojového učenia (ML) do bioinformatických nástrojov a pracovných postupov prináša revolúciu v tejto oblasti. AI a ML sa používajú na analýzu veľkých súborov údajov, predpovedanie proteínových štruktúr a vývoj nových liekov.
Aký typ biologických údajov možno použiť v bioinformatike?
Pokrok v molekulárnej biológii spojený so zlepšením výpočtových zdrojov teda viedol k vzniku veľkého množstva biologických údajov a prístupov k analýze údajov, ktoré možno použiť na urýchlenie vývoja v biotechnológii. Spoločne sú rozsiahle biologické údaje a súvisiace bioinformatické analýzy známe pod príponou „-omika“ a zahŕňajú genomiku, fylogenomiku, transkriptomiku, proteomiku, metabolomiku a metagenomiku.
Analýza proteínovej a nukleotidovej sekvencie - práca so sekvenačnými dátami
Nomenklatúrny systém, ktorý používame v práci v oblasti bioinformatiky, je založený na odporúčaniach Medzinárodnej únie čistej a aplikovanej chémie (IUPAC). Je užitočné dodržiavať tento nomenklatúrny systém, aby bolo možné ľahko a jednotne porovnávať súbory údajov z rôznych laboratórií nachádzajúcich sa po celom svete. Databázové inštitúcie a časopisy, ktoré publikujú výskumné správy, prísne dodržiavajú tieto odporúčania, aby zabezpečili jednotnosť a pomohli rýchlej reprodukovateľnosti. V tejto časti si prejdeme základný nomenklatúrny systém pre nukleové kyseliny a bielkoviny.
Aminokyseliny slúžia ako základné jednotky na stavbu bielkovín v živých organizmoch. Zatiaľ čo v prírode existuje viac ako 500 rôznych typov aminokyselín, ľudský genetický kód špecificky kóduje iba 20. Každá aminokyselina má jedinečnú štruktúru, ktorá určuje jej vlastnosti a úlohu pri syntéze bielkovín. Na reprezentáciu 20 rôznych aminokyselín (kodogénnych), ktoré sa podieľajú na stavbe bielkovín bol vytvorený systém kódov s dvoma bežne používanými formátmi: jednopísmenové a trojpísmenové kódy.
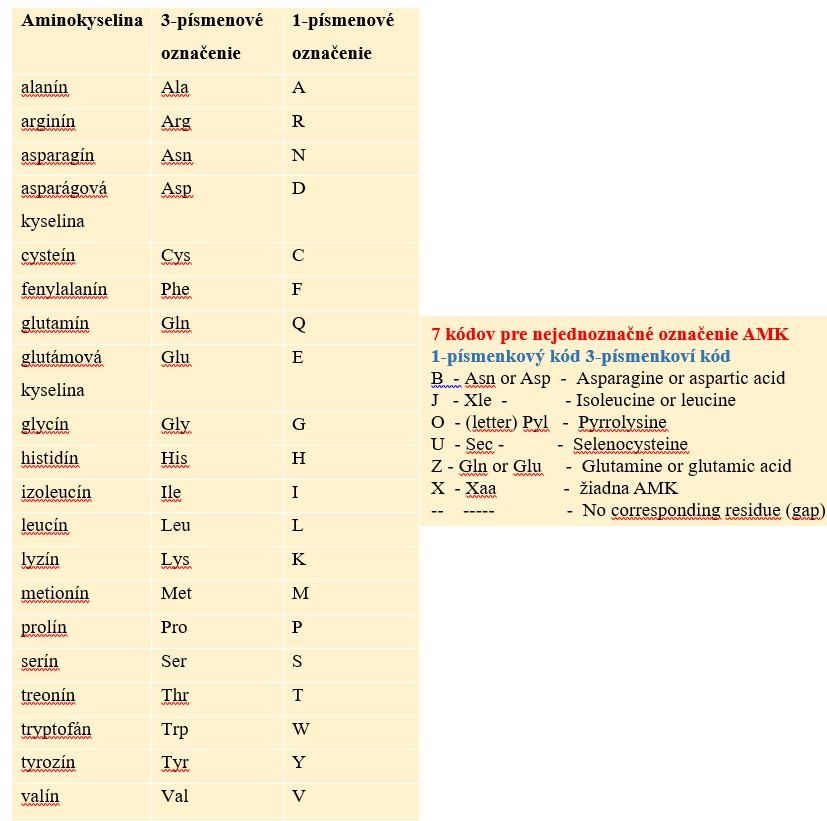
IUPAC kód (International Union of Pure and Applied Chemistry) pre aminokyseliny
* B a Z - nemožnosť rozlíšiť medzi Asp a Asn (alebo Glu a Gln)* J - nemožnosť rozlíšiť medzi Ile a Leu pomocou hmotnostnej spektrometrie, či sekvenovania.
* Vzácne aminokyseliny Pyl (Pyrrolysine) a Sec (selenocysteine) sú špecifikované stop kodónmi UAG (Pyl) a UGA (Sec) čítanými v špecifickom kontexte.
* Kód X sa stále často používa ako zástupné písmeno, keď nepoznáte aminokyselinu na danej pozícii v sekvencii.
* Zarovnávacie programy používajú „-“ na označenie pozícií zjavne chýbajúcich v sekvencii.
Nukleové kyseliny (DNA, RNA) sú makromolekuly zložené z opakujúcich sa podjednotiek nazývaných - nukleotidov. ktorý obsahuje 5-uhlíkový cukor (pentóza: deoxyribóza príp. ribóza), dusíkatú bázu a fosfátovú skupinu. Medzi dusíkaté bázy tvoriace DNA patrí : adenín, guanín, tymín, cytozín a medzi dusíkaté bázy tvoriace RNA patrí adenín, guanín, cytozín a uracil.
Prvé 4 bázy G,A,T,C, ich symboly ako základ nomenklatúry sú jasné. Pri určovaní sekvenčných údajov prostredníctvom experimentov niekedy nemusí byť identita sekvencie v konkrétnej polohe jasne identifikovateľná v dôsledku kompresných artefaktov alebo iných problémov súvisiacich so sekundárnou štruktúrou. Vo väčšine prípadov je možné problém vyriešiť opakovaním experimentu a tiež sekvenovaním komplementárneho reťazca. V niekoľkých prípadoch môžu pretrvávať nejasnosti.
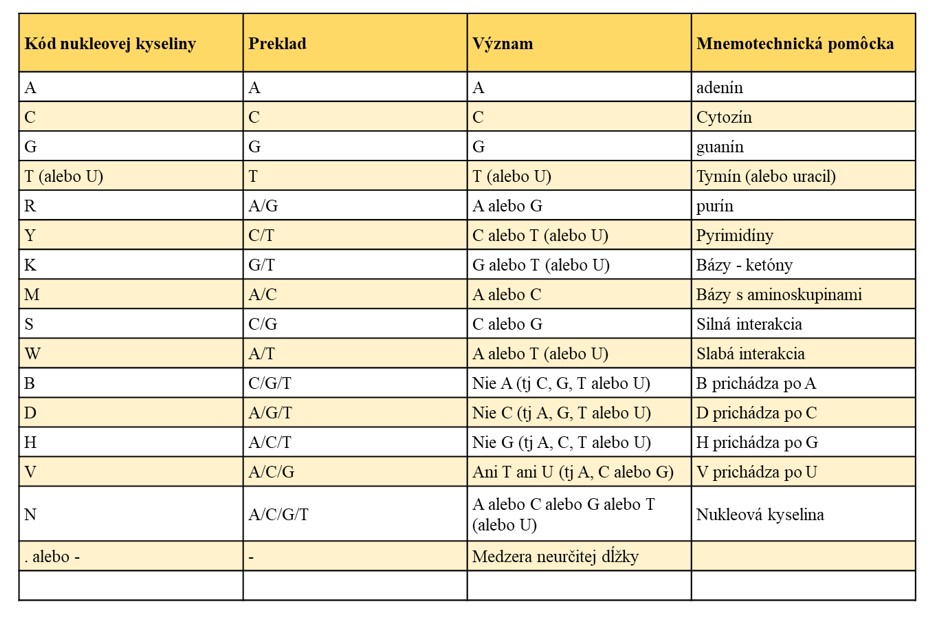
IUPAC kód (International Union of Pure and Applied Chemistry) pre nukleové kyseliny.
Túto tému nájdeš aj
v e-learningovom kurze
Prihlás sa do e-learningového kurzu a okrem plnej verzie textovej časti tejto témy získaj prístup aj k:
- prezentáciám tejto témy
- podporným materiálom k tejto téme
- možnosti otestovať svoje vedomosti
- komunikácii s autormi tejto témy
- diskusnému fóru k tejto téme
Ak ešte nemáš prístup k e-learningovému kurzu, prečítaj si, ako ho môžeš získať.

