Basic Local Alignment Search Tool (BLAST) je program na vyhľadávanie podobnosti sekvencií, ktorý je možné použiť cez webové rozhranie alebo ako samostatný nástroj.
BLAST je široko používaný bioinformatický program, ktorý prvýkrát predstavil Stephen Altschul a kolektív v roku 1990 a odvtedy sa stal jedným z najpopulárnejších nástrojov na vyhľadávanie podobnosti sekvencií. Tento počítačový algoritmus sa zameriava na porovnávanie aminokyselinových sekvencií rôznych proteínov alebo nukleotidových sekvencií nukleových kyselín pričom vyhľadáva homológne sekvencie vo vopred definovaných a anotovaných databázach podľa výberu používateľa. BLAST je k dispozícii na použitie online na webovej stránke Národného centra pre biotechnologické informácie (NCBI) (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
Vďaka dostupnosti sekvenovacích technológií sú k dispozícii veľké databázy sekvencií s možnosťou hľadať, či sa niektoré sekvencie alebo ich časti zhodujú alebo aspoň podobajú. Podobnosti medzi sekvenciami v databázach pochádzajú buď z toho, že sa viackrát sekvenovala tá istá sekvencia, alebo že sa dve sekvencie vyvinuli mutáciami zo sekvencie v spoločnom predkovi. Takéto sekvencie sa nazývajú homologické a pod pojmom hľadanie homológov (homology search) sa myslí hľadanie takých podobností medzi sekvenciami, ktoré s veľkou pravdepodobnosťou vznikli práve zdieľanou evolučnou históriou. Nájdené podobnosti väčšinou zobrazujeme vo forme zarovnania (sequence alignment). Každý riadok zarovnania je jedna zo sekvencií, pričom sa medzi ne pridávajú pomlčky tak, aby boli oba riadky rovnako dlhé a aby v stĺpcoch pod sebou boli čo najčastejšie rovnaké bázy. Skupinu susedných pomlčiek v zarovnaní nazývame medzera (gap).
Zoradenie sekvencií je v bioinformatike kľúčové z niekoľkých dôvodov:
• Homológia: Pomáha určiť, či sú dve sekvencie evolučne príbuzné, čo naznačuje spoločného predka. To je nevyhnutné pre pochopenie evolučných vzťahov, funkcie a štruktúry proteínov .
• Funkčná anotácia: Porovnaním novo sekvenovaného proteínu so známymi proteínmi môžu výskumníci odvodiť jeho funkciu na základe konzervovaných oblastí.
• Fylogenetická analýza: Zarovnanie sekvencií z rôznych druhov pomáha rekonštruovať evolučné stromy, odhaľuje evolučnú históriu a príbuznosť organizmov.
• Analýza mutácií: Identifikuje mutácie, inzercie, delécie a preskupenia v sekvenciách a poskytuje pohľad na genetické variácie a ich dôsledky.
• Objav liečiv: Zoradenie sekvencií pomáha pri identifikácii potenciálnych cieľov liečiv porovnaním proteínových sekvencií patogénov s hostiteľskými organizmami.
• Anotácia genómu: Pomáha anotovať genómy zarovnaním sekvencií DNA alebo RNA so známymi funkčnými prvkami, ako sú gény a regulačné oblasti.
• Štrukturálna biológia: Zarovnanie proteínových sekvencií môže viesť k predikcii proteínových štruktúr a identifikácii konzervovaných štruktúrnych motívov.
• Metagenomika: Zarovnanie sekvencií zo vzoriek prostredia pomáha identifikovať mikrobiálne druhy a pochopiť štruktúru mikrobiálnych spoločenstiev.
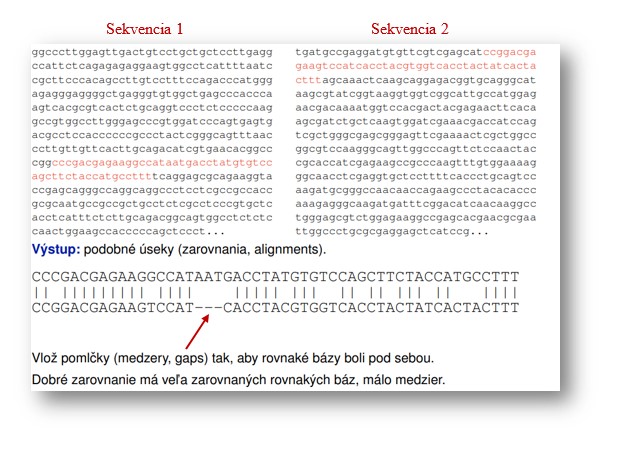
Dve sekvencie a ich lokálne zarovnanie (Zdroj: Brejová, https://compbio.fmph.uniba.sk/vyuka/mbi/images/d/dd/P-aln1.pdf)
Existuje päť typov (variantov) BLAST, ktoré sú diferencované na základe typu sekvencie (DNA alebo proteín) dopytovaných databázových sekvencií.
BLASTN porovnáva dopytovanú sekvenciu nukleotidov s databázou sekvencií nukleotidov.
BLASTP porovnáva proteínovú dotazovaciu sekvenciu s databázou proteínovej sekvencie.
BLASTX porovnáva dopytovanú nukleotidovú sekvenciu s databázou proteínových sekvencií tým, že prekladá nukleotid na proteín a hľadá ho v proteínových databázach.
TBLASTN porovnáva proteínovú dotazovaciu sekvenciu s databázou nukleotidových sekvencií preložením nukleotidových sekvencií a ich porovnaním s proteínovou sekvenciou.
TBLASTX porovnáva dopytovanú nukleotidovú sekvenciu s databázou nukleotidových sekvencií prekladom skúmanej sekvencie do všetkých šiestich čítacích rámcov a ich porovnávaním s nukleotidovými sekvenciami.
BLAST VYHĽADÁVANIE
V prípade úhynu zvieraťa sa realizuje pitva v čo možno najkratšom čase a súčasne sa odoberajú vzorky pre ďalšie laboratórne vyšetrenie. Po vonkajšom obhliadnutí sa prevádza vyšetrenie podkožného tkaniva, svaloviny, kostí, telových dutín a vnútorných orgánov. Zvláštna pozornosť sa venuje tráviacemu traktu. Posudzuje sa stav seróz, naplnenie žalúdka a čriev. Po otvorení hrvoľa, žalúdka alebo predžalúdka možno zistiť zápach, charakterizujúci niektoré toxické látky (napr. zápach po cesnaku pri otrave fosfidom zinku). Rovnako je potrebné posúdiť zloženie a sfarbenie žalúdočného obsahu, prípadne prítomnosť podozrivých predmetov v žalúdku (napr. sfarbené rodenticídne nástrahy, zvyšky nástrahového zrna). Z vnútorných orgánov je potrebné zvlášť starostlivo preskúmať pečeň, obličky a slezinu.
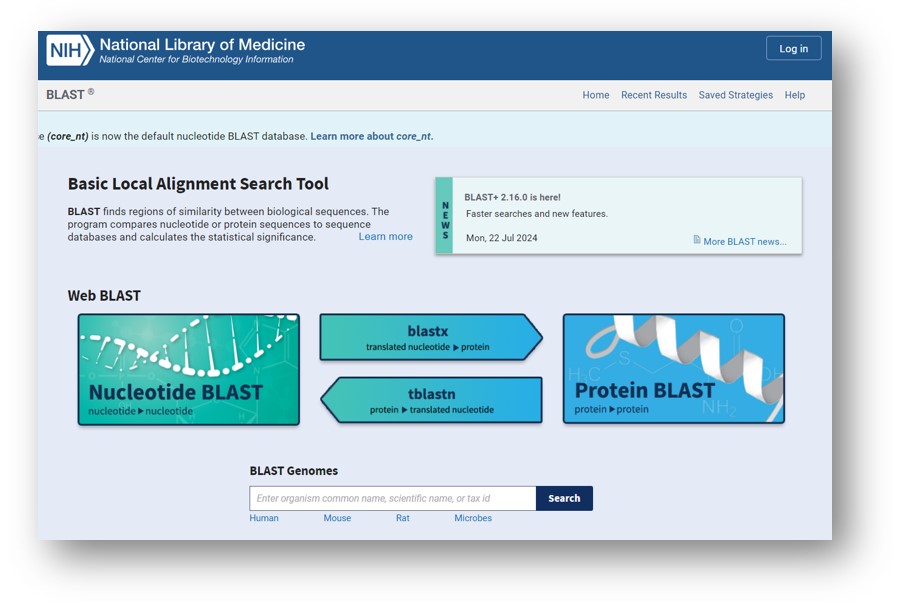
Domovská stránka BLAST v NCBI.
Ako môžeme vidieť na hlavnej strane, BLAST má v súčasnosti veľa možností. Ak je cieľom porovnať sekvencie z jedného z dostupných publikovaných genómov, potom by sme vybrali genómy BLAST Assembled RefSeq. Za zmienku stojí najmä pridanie mnohých špecializovaných nástrojov BLAST, vrátane Primer-BLAST, ktoré by vám pomohli navrhnúť alebo skontrolovať PCR primery. Vo všeobecnosti však budeme skôr vyberať z programov Basic BLAST. Napríklad, ak pracujeme s DNA, vyberte blastn (nukleotidový blast). Ak pracujete s proteínovými sekvenciami, potom použite blastp (proteínový blast). Avšak bez ohľadu na zvolený BLAST sú základy dosť podobné.
Po vložení dopytovacej sekvencie, prípadne nahratia zvolenej sekvencie pomocou „Choose File“ prejdeme do časti „Choose Search Set“/Výber programu“ posúvaním stránky nadol.
Vyberieme program na optimalizáciu vyhľadávania a kliknutím na tlačidlo BLAST spustite vyhľadávanie. Predvolenou databázou je „Core nucleotide database (core_nt)“, ktorá umožňuje rýchlejšie vyhľadávanie nukleotidov BLAST s presnejšími výsledkami.
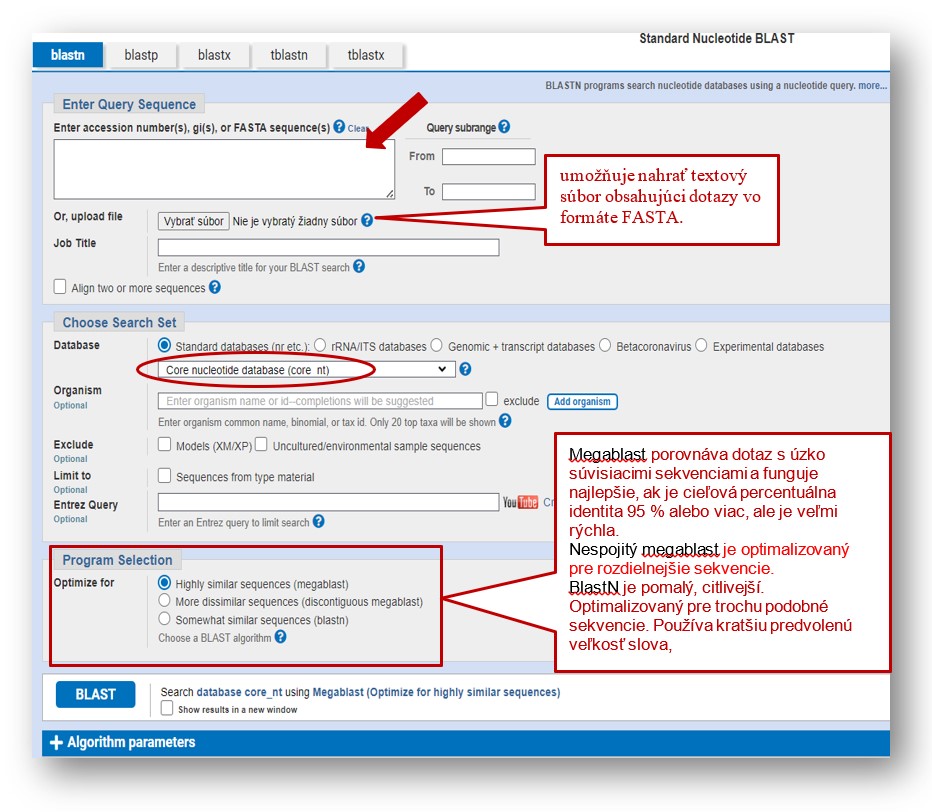
Hlavná stránka BLAST pre vyhľadanie a porovnanie sekvencií.
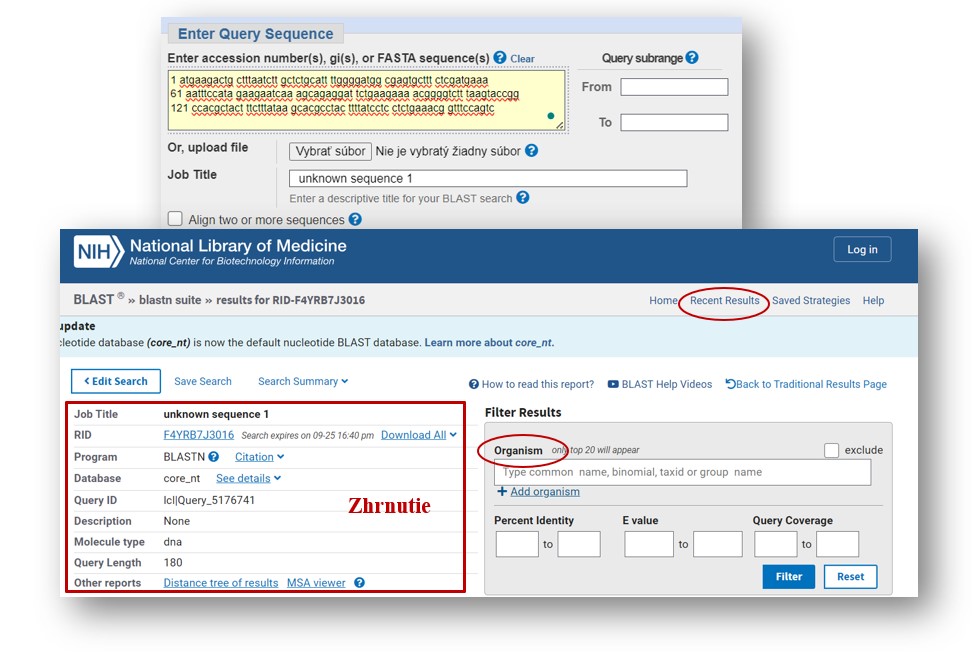
Karta so zobrazením výsledkov BLAST pre dopytovanú sekvenciu.
V prvej časti karty so zobrazenými výsledkami sa objaví krátke zhrnutie o zadanej sekvencii. Zhrnutie poskytuje informácie o rôznych aspektoch vyhľadávania (obrázok 119). RID predstavuje jedinečný identifikátor priradený tomuto vyhľadávaniu na účely zdieľania a hlásenia problémov. V políčku Download All (Stiahnuť všetko) sa zobrazí zoznam možností na uloženie kompletného výsledku vyhľadávania v požadovanom formáte, napríklad v novom formáte XML (XML2), JSON a CSV.
• Database (Databáza) uvádza názov cieľovej databázy, v ktorej sa vyhľadáva.
• Query ID (ID dotazu) je poradové číslo dotazu, pre ktorý sa zobrazuje výsledok. Ak sa používa accession (prístup), je prepojený so záznamom v databáze NCBI.
• Description (Popis) je názov sekvencie dotazu z jej defline FASTA.
• Molecule type (Typ molekuly) zobrazuje typ sekvencie.
• Query length (Dĺžka dotazu) je dĺžka aktuálneho dotazu.
• Other reports (Iné zostavy) uvádza odkazy na iné formáty zostáv, ktoré nie sú integrované do nového zobrazenia na karte.
Využi pri ďalšom štúdiu tejto témy e-learningový kurz.
Túto tému nájdeš aj
v e-learningovom kurze
Prihlás sa do e-learningového kurzu a okrem plnej verzie textovej časti tejto témy získaj prístup aj k:
- prezentáciám tejto témy
- podporným materiálom k tejto téme
- možnosti otestovať svoje vedomosti
- komunikácii s autormi tejto témy
- diskusnému fóru k tejto téme
Ak ešte nemáš prístup k e-learningovému kurzu, prečítaj si, ako ho môžeš získať.
